亲水作用色谱法测定甜菊糖主要极性组分
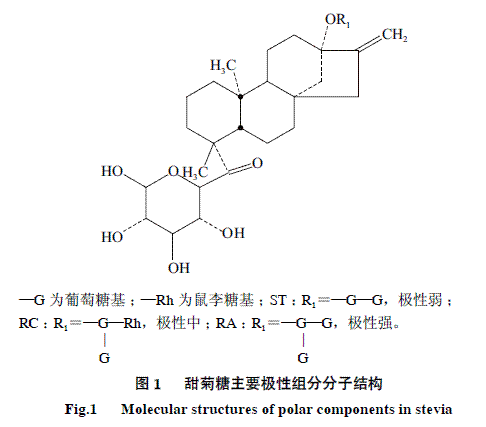
甜菊糖是从甜叶菊中提取出的一种天然甜味剂,具有高甜度、低热量、安全无毒等特点, 广泛应用于食品、饮料、酿酒、医药、日用化工等行业,特别适宜肥胖症、糖尿病、高血压、动脉硬化、龋齿患者食用[1-3]。甜菊糖含有8种糖甙成分,具有经济价值且含量较多的主要是莱鲍迪甙A(rebaudioside A, RA)、甜菊糖甙(stevioside,ST)、莱鲍迪甙C(rebaudioside C,RC)。RA的甜度为蔗糖 的350~450倍,口味纯正;ST的甜度为蔗糖的150~250倍,但略带苦味;RC的苦味较重,严重影响甜菊糖的品质[4-5]。它们之间的价格也相差数倍,故准确定量甜菊糖样品中三者的含量尤为重要。ST、RC和RA的分子结构如图1所示,其主要区别仅在于糖基部分葡萄糖的数目以及有无鼠李糖,这就使它们的分离与检测变得十分困难。
目前常用的检测方法有毛细管电泳( c a p i l l a r ye l e c t r o p h o r e s i s ,CE)法、薄层色谱( t h i n l a y e rc h r o m a t o g r a p h y ,TLC)法、高效液相色谱( h i g hperformance liquid chromatography,HPLC)法等[6-9],其中HPLC以其精准简便而被广泛采用[10]。Ludmila[9]、Prakash[11]、吴岩[12]、王瑞[13]等分别使用C18柱与CN柱对甜菊糖样品进行了色谱分离,但分离度均较差,定量分析不准确。GB 8270—1999《食品添加剂:甜菊糖甙》采用NH2柱[10]。刘超[14]、Kolb[15]、Liu Yongfeng[16]等使用NH2柱对甜菊糖样品中的RA、RC和ST进行了分离检测,该法分离效果较C18柱和CN柱虽有较大提高,但分离效果仍然不理想,尤其RC分离较差,导致各组分色谱纯度的准确性欠佳。
亲水作用色谱(hydrophilic interaction liquidchromatography,HILIC)是一种以极性固定相,水-极性有机溶剂为流动相的色谱模式。它与正相色谱相似,但以水为强洗脱剂,尤其适用于反相色谱中保留不完全或不保留极性组分的分离[17-18]。目前尚未见使用强碱性阴离子色谱柱(strong base anion exchange,SAX)对甜菊糖各组分进行分离检测的报道,本实验根据HILIC模式建立高效液相色谱-强碱性阴离子色谱柱-紫外检测器(hydrophilic interactionliquid chromatography-strong base anion exchange-ultravioletdetector,HPLC-SAX-UVD)的检测方法,并与GB 8270—1999的NH2柱方法进行比较,以期克服原HPLC检测方法的一些弊端,使检测结果更为精确。
1· 材料与方法
1.1 材料与试剂
甜菊糖样品(RA、ST、RC质量分数分别为60%、14%、8%) 山东曲阜圣仁制药有限公司;乙腈(色谱纯)美国霍尼韦尔公司;超纯水 自制;除另有规定外,所用试剂均为分析纯。
1.2 仪器与设备
LC-20AT高效液相色谱仪(配有SPD-M20A紫外(ultraviolet,UV)检测器、LTⅡ蒸发光散射检测器(evaporative light-scattering detector,ELSD)、CTO-20A柱温箱)、Inertsil NH2柱(250mm×4.6mm,5μm) 日本岛津公司;Hypersil SAX强碱性阴离子色谱柱 美国热电公司;超纯水仪 上海优普实业有限公司;GA-3000型低噪音空气泵 北京中兴汇利科技发展有限公司。
1.3 方法
1.3.1 色谱条件
色谱柱: H y p e r s i l S A X 强碱性阴离子柱(250mm×4.6mm,5μm);流动相:乙腈-水(85:15,V/V);流速:2.0mL/min;柱温:30℃;进样量:5μL;检测器:紫外检测器,检测波长210nm。
GB 8270—1999色谱条件[19],色谱柱:NH2基柱;流动相:乙腈-水(80:20,V/V);流速:1.2mL/min;进样量:15μL;检测器:紫外检测器,检测波长210nm。
1.3.2 标准曲线溶液的配制
准确称取甜菊糖样品4.000g于50mL烧杯中,用超纯水溶解、转移至100mL容量瓶中定容,得标准母液。分别量取标准母液0.1、1.0、3.0、5.0、6.0、8.0、10.0mL于100mL容量瓶中,并用超纯水定容,配制成一系列质量浓度的标准液,3种组分的质量浓度分别为ST 0.056、0.560、1.680、2.800、3.360、4.480、5.600mg/mL,RC 0.032、0.320、0.960、1.600、1.920、2.560、3.200mg/mL,RA 0.240、2.400、7.200、12.000、14.400、19.200、24.000mg/mL,并用微孔滤膜(0.45μm)过滤备用。
2 ·结果与分析
2.1 色谱条件的选择
2.1.1 流动相和检测器的选择
以乙腈-水为流动相,采用UV检测器和ELSD串联的方式,对同一样品溶液进行比较。调节乙腈-水的比例,对同一个样品进行检测,得到一条符合HILIC特征的“U”型曲线(图2)。由图2可知,当乙腈体积分数为70%以下时,各组分基本没有实现分离,而在80%以上,ST、RC、RA按极性递增的顺序依次被洗脱,分离效果明显提高,并且各组分对水含量的变化尤为敏感。因此,进一步以80:20、82:18、85:15、87:13(V/V)的乙腈-水作为流动相,对同一个样品进行检测,得到图3的结果。由图3可知,随着乙腈体积分数的增大,各物质的分离度明显提高,当乙腈体积分数达到85%时,ST、RC、RA均达到较好的基线分离。85%乙腈之后,ST,RC,RA的响应值明显下降,且各组分的保留时间随乙腈体积分数的增大呈指数型增长。上述实验特征表明,该体系完全符合HILIC的分离模式。
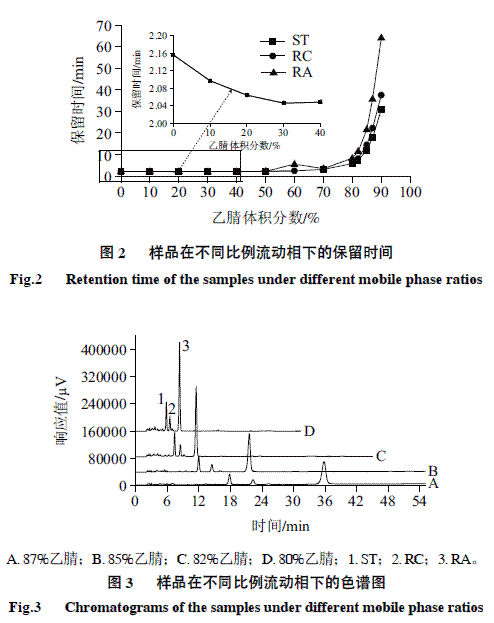
此外对同一样品,UV(210nm)响应值比ELSD(压力360kPa、增益1、温度30℃)高出8~60倍。综合考虑分离度、保留时间和检测灵敏度,选用乙腈-水(85:15,V/V)作为流动相,UV检测器为目标检测器。
2.1.2 流速的选择
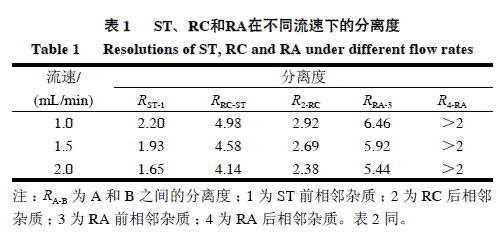
由表1可知,在不同流速下,主要组分均达到了完全分离;当流速为2.0mL/min时,3个主要成分ST、RC、RA的R值均能达到1.5以上,且峰的对称性很好。综合考虑R值、检测时间和柱压,最终选择流速为2.0mL/min。
2.1.3 柱温的选择
考察了30℃和35℃条件下样品在SAX柱上的分离情况。结果显示,柱温对各组分的R值、保留时间以及响应值的影响甚微。故选用柱温30℃。
2.2 本方法和GB 8270—1999法的比较
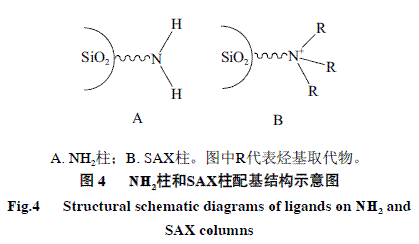
本法采用的SAX柱与国标法采用的NH2柱相比,均以硅胶为基质;不同的是前者以季铵基(—N+(R)3)为配基,后者则以伯胺基(—NH2)为配基,两种配基都具有亲水性,但极性有所不同(图4);采用的流动相均为一定比例的乙腈-水溶液,故两者同为HILIC模式。通过SAX柱和NH2柱最优条件下分离效果的对比(图5),发现甜菊糖在SAX柱上的杂质峰数目要多于NH2柱的,且主要组分与杂质之间的分离度也明显优于NH2柱(表2)。
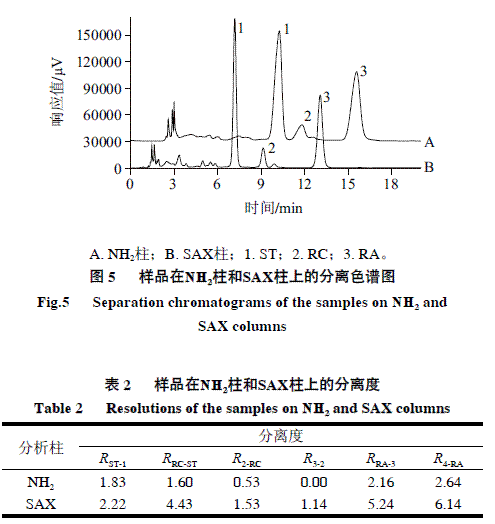
目前对HILIC的分离机理还存在争议,通常认为与静电、氢键和偶极作用等相关[14,20-24]。本实验中甜菊糖属于非电离物质,无法进行离子交换,故与色谱柱之间无静电作用。因此,ST、RC和RA在色谱柱上的分离可能是由于离子-偶极和氢键的协同作用。离子-偶极作用是指带电离子和偶极分子之间的作用。离子交换填料本身含有带电离子基团,甜菊糖极性组分自身具有一定的偶极矩,因此填料与ST、RC和RA间可能存在着离子-偶极作用。图6以RA为例,给出了甜菊糖在SAX柱上的作用机理模型。
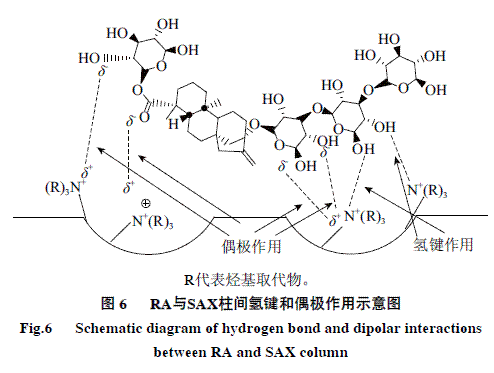
由于ST、RC和RA组分的极性差异,使三者的偶极矩大小不一致,从而导致三者与SAX柱上季铵基间形成的离子-偶极作用不一样。与NH2柱相比,这种差异在SAX柱上体现得更为显著,这可能是由于季铵基的极性明显强于伯胺基。此外,季铵基N上的H已全部被烷基取代,无法形成分子间氢键,而其连有更多的烷基可以提供更多的电子,使N原子的电子云密度增大,有利于与甜菊糖分子的羟基形成氢键。季铵基与ST、RC和RA形成氢键的强弱有所不同,从而造成了SAX柱对三者吸附选择性的差别。而伯胺基含有未取代的H,易形成分子间的氢键,不利于与甜菊糖形成氢键,因此对ST、RC和RA的选择性差异不大。综上所述,甜菊糖极性组分在SAX和NH2柱上的保留可能均与离子-偶极和氢键的协同作用相关。然而与弱极性的NH2柱相比,ST、RC和RA在强极性SAX色谱柱上的差异性表现的更为显著。因此,甜菊糖极性组分在SAX柱上的分离与检测显然要优于NH2柱。
2.3 标准曲线及最低检出限

用所建立的HPLC-SAX-UVD方法对所配标准溶液进行检测,以各物质峰面积A为纵坐标,各物质质量浓度(C)为横坐标,用最小二乘法进行线性回归,得到各自标准曲线。由表3可知,ST、RC和RA的线性范围较宽,且相关性优良。按信噪比RSN≥3推算,ST、RC和RA的最低检出限分别为5.0、5.9μg/mL和6.0μg/mL。
2.4 方法的准确度和精密度
为考察方法的准确度和精密度,在线性范围内按照低、中、高3个水平加入甜菊糖样品。按色谱条件进行定量分析,计算方法的回收率和标准偏差,结果见表4。
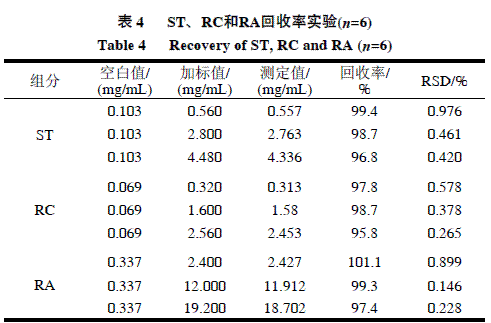
由表4可知,在所测范围内,ST、RC、RA的加标回收率为95.8%~101.1%,表明该方法准确度好;RSD均小于1.00%,故所建立的方法具有较高的精密度。
3·结 论
本实验根据HILIC分离模式建立了HPLC-SAX-UVD检测方法,首次引入强碱性阴离子色谱柱对甜菊糖极性组分进行分离,克服了国标法分离效果不佳、分离度较差的弊端。初步探讨了HILIC的分离机理,并推断产生这一机理的诱因可能为氢键、偶极作用等。通过方法学考察,证明该方法简单、灵敏、准确且重现性好,适用于甜菊糖主要组分ST、RC和RA含量的精确测定,为甜菊糖样品的准确检测提供了可靠的方法。